年間100万人に1人いるかどうかの極めて稀な脳疾患の1つにプリオン病という病気があります。
その中でもクロイツフェルト・ヤコブ病(Creutzfeldt-Jakob)がヒトプリオン病の80%を占め、脳神経細胞の機能が障害されます。
今回は、このプリオン病について
- 症状
- 原因
- 診断
- 治療法
を説明致します。
プリオン病とは?
中枢神経に存在する正常なプリオン蛋白が異常プリオン蛋白に変性し、分解されないままに脳内に蓄積され、脳神経細胞の機能が障害されるために起こる脳疾患です。
好発年齢として、50~60代の初老期に多く、中でも若干女性に多く現れます。

プリオン病の症状は?
進行性疾患なため、発症時から急激に症状は悪化していきます。
初期症状
- 精神症状
- 視覚異常
- 歩行障害
などがあり、健忘症や抑うつなど、運動失調症状が現れ、異変に気づきます。
進行症状
進行する認知症が主な症状で、言葉が出にくくなり、自発語不能となるまで急速です。四股のミオクローヌス症状も現れます。
末期症状
ミオクローヌス症状は消失するも、無動性無言から除皮質硬直や屈曲拘縮に発病後数カ月以内に進展します。
また、全身が衰弱し、呼吸が困難となっていき、肺炎などの感染症を合併し、発症から1〜2年の短い間で死に至ります。
プリオン病の原因は?
ウイルスや細菌が原因ではなく、プリオンという感染因子が原因です。
感染型のプリオン蛋白が正常なプリオン蛋白を変性させることにより、中枢神経に蓄積、神経細胞を死滅させることが原因と考えられています。
- 角膜移植
- 脳硬膜移植
- 牛海綿状脳症(狂牛病)
などが関係するともいわれています。
プリオン病の分類
孤発性、遺伝性、感染性とに分けられ、さらに孤発性は、孤発性Creutzfeldt-Jakobに、遺伝性は家族性Creutzfeldt-JakobとGerstmann-Straussler-Scheinker病、致死性家族不眠症に、感染性はKuru斑、医原性Creutzfeldt-Jakob、特異性Creutzfeldt-Jakobとに分けられます。
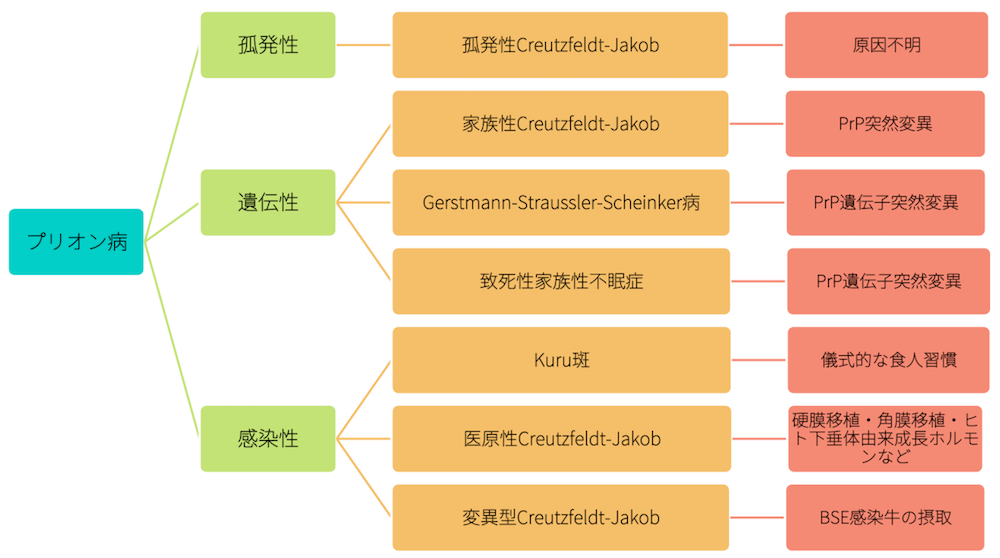
プリオン病の中でも最も患者数が多いのは孤発性です。遺伝性では染色体優位遺伝を示します。
また、儀式的な食人習慣は禁止されたことにより、発病は終息しました。
また移植では、現在ヒト由来成分を極力使用しないようにし、十分に不活性を行っています。
但し、変異性Creutzfeldt-Jakobの場合好発年齢が異なります。
平均29歳とされ、また1990年代半ばと比較的最近出現した新病型です。
プリオン病の診断は?
- 脳脊髄液検査
- MRI検査
- 脳波検査
脳脊髄液検査
髄液内に神経特異性エノラーゼ、14-3-3蛋白、総タウ蛋白の上昇が見られます。
MRI検査
特に拡散強調像やFLAIR像が有用です。
孤発性Creutzfeldt-Jakobでは大脳皮質に、変異型Creutzfeldt-Jakobでは基底核と視床に高信号域が確認出来ます。
また、病態が進行すると脳の萎縮がはっきりと分かります。
症例 70歳代男性 進行する認知症
頭部MRI 拡散強調像
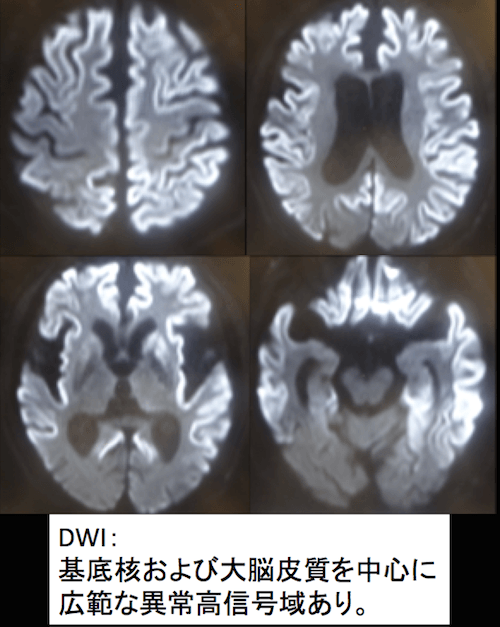
拡散強調像で両側の基底核及び大脳皮質を中心に、広範な異常な高信号を認めています。
CJDを疑う所見です。
脳波検査
全誘導にわたって、高振幅鋭波を伴う周期性同期性放電が認められます。
プリオン病の治療法は?
有効な治療法は見つかっていません。
そのため、発症すると1~2年で死亡するとされています。
最後に
- プリオン病は進行性疾患で、発症から1~2年で死亡する
- 進行する認知症が主な症状で、急激に症状は悪化していく
- プリオン病は、孤発性、遺伝性、感染性に分類される
- 脳脊髄液検査・MRI検査・脳波検査によって診断される
- 有効な治療法はない
プリオン病診断後は、他人への血液感染リスクを避けるため、不用意な採血や献血は行えません。
若年性で急速に進展する認知症状がある場合、詳しい検査をお勧めします。